
苏州德锐特开源软件FEP-SPell-ABFE
正式发布在美国化学会旗下JCIM期刊
结合自由能的计算,尤其是针对蛋白-配体复合物体系,是计算化学领域的一个重要研究方向。炼金术自由能模拟(Alchemical Free Energy Simulations)技术被业内认为是计算结合自由能最精确的方法。该技术通过在始末热力学态之间增加一系列虚拟的中间态,从而实现缓慢的演变。
绝对结合自由能ABFE(Absolute Binding Free Energy)计算方法通过双解耦(double-decoupling)将配体分子转变成不会被周围环境(溶剂化环境和结合位点)所感知的虚拟分子,评估配体与靶蛋白之间的结合自由能。与相对结合自由能(RBFE)不同,ABFE计算不依赖于配体之间的结构相似性,因此适用于更广泛的分子自由能计算场景。然而,与RBFE相比,计算ABFE还需要额外的步骤,如引入或消除蛋白质和配体之间的约束,这使模拟协议变得复杂,并增加了计算需求。
近日,苏州德锐特公司联合华东师范大学梅晔教授课题组在《Journal of Chemical Information and Modeling》期刊上正式发布了共同研发的自动化预测ABFE的计算工具FEP-SPell-ABFE(https://pubs.acs.org/doi/10.1021/acs.jcim.4c01986),该工具提供了一套高效、自动化的ABFE计算流程,能够轻量化地部署在高性能计算集群上,为结构多样的配体分子提供了更加灵活、精准的结合自由能预测方案。
FEP-SPell-ABFE 是一款基于 Python 的开源自动化工作流,专为计算药物分子与靶蛋白的绝对结合自由能(ABFE)设计。该工具大幅降低了计算准备的工作量,能够简单快速地预测候选分子的结合亲和力。操作简便,用户仅需准备三个关键文件即可开展计算:
- 蛋白受体结构文件(PDB格式)
- 候选配体分子文件(SDF格式)
- 管理工作流和分子动力学模拟参数的配置文件(yaml)
用户可根据需求灵活调整配置文件config.yaml,通常只需简单修改相关配置,即可一键提交任务(参见后文流程示例),程序将自动执行整个模拟流程,并生成包含结合自由能数据的CSV文件。此外,该工具依托 SLURM 计算资源管理系统,可实现跨模块的自动化任务调度与计算资源分配,支持在高性能计算集群上对大批量配体分子并行计算,能够大幅提升计算效率。
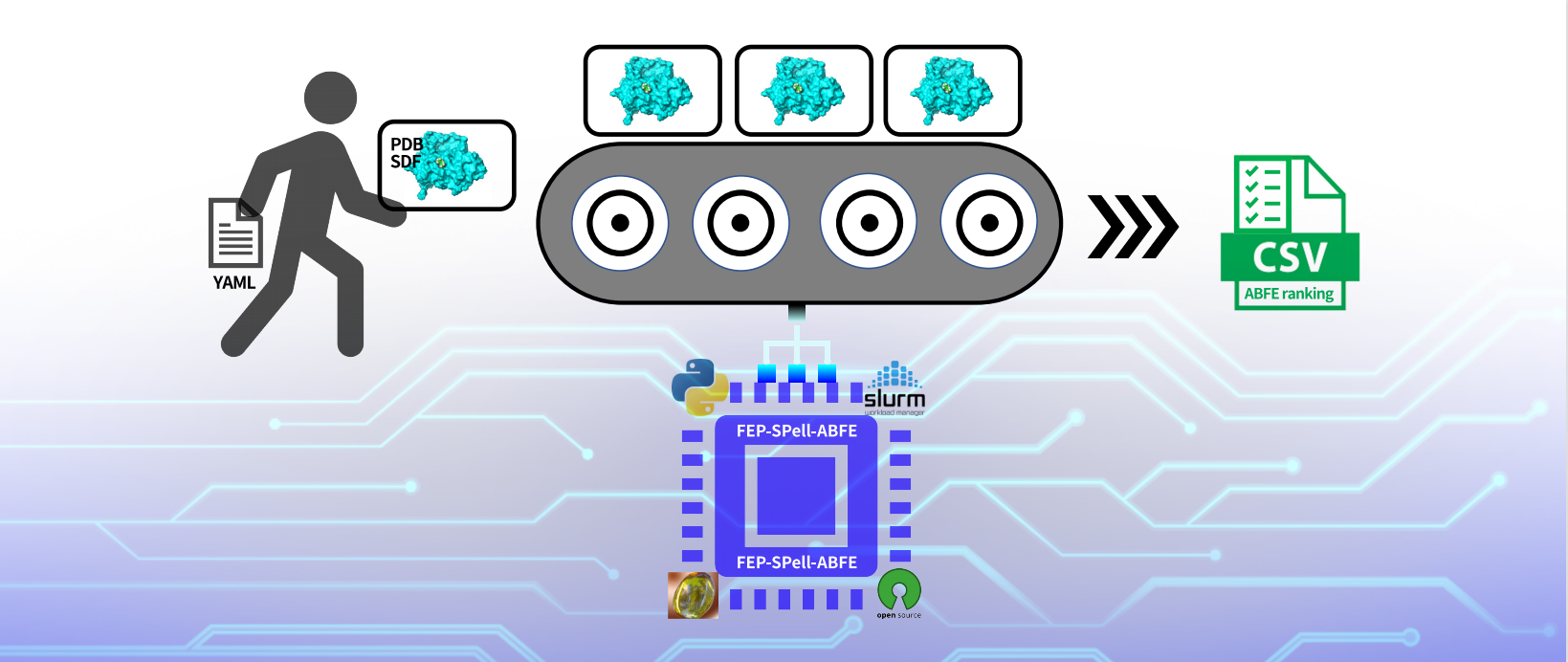
🚀 核心优势
✅ 自动化流程:包含体系构建、分子动力学平衡、自由能计算及结果分析的完整流程,极大减少用户干预,实现较高程度的自动化。
✅ 高精度预测:基于严格的热力学循环,以AMBER软件作为底层分子动力学模拟引擎,提供可靠的结合自由能计算结果。
✅ 高效率批量计算:支持SLURM计算资源管理系统,实现在高性能计算集群上同时运行大批量配体的计算任务,并行处理较大规模药物筛选任务。
✅ 开源可拓展:用Python开发,代码开源,支持用户根据具体研究需求进行优化和定制,灵活适应不同计算场景下的科学问题。
📌技术内核:ABFE热力学循环:
通过一系列热力学变换,将配体分子从水相中的自由状态转变为与蛋白质靶标结合的复合物,累加过程中的自由能变获得该小分子与靶标蛋白的绝对结合自由能ΔGbind。
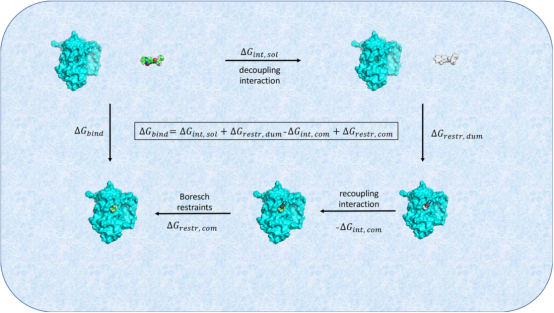
- ΔGint,sol→ 在溶液中配体的去耦合(Decoupling in Solution)
- 关闭配体在水溶液中的范德华力和静电相互作用,使其从溶液“消失”变成理想气体状态。
- 计算模拟这一去耦合过程中自由能的变化。
- ΔGrestr,dum→ 对小分子与蛋白的相对位置施加约束(Turning on Restraints)
- 在去耦合的状态下,对配体施加Boresch 约束,固定其在结合位点的空间位置和取向。
- 这部分的自由能变化可以通过解析计算获得,无需额外模拟。
- ΔGint,com→ 在靶蛋白的结合口袋中配体的重耦合(Recoupling in Binding Site)
- 让配体在蛋白结合位点重新恢复范德华和静电相互作用,模拟其从无相互作用状态变为完全结合状态。
- 计算模拟这一耦合过程中自由能的变化。
- ΔGrestr,com→ 去除约束力(Turning off Restraints)
- 解除Boresch 约束,允许配体能够在结合位点自由运动,恢复其真实状态。
- 计算模拟去除约束力过程的自由能变化。
通过上述四步计算出体系中不同相互作用的自由能变化,最终以获得绝对结合自由能,用于评估配体和蛋白的结合强度。
- 计算流程:
- Topology Construction:生成模拟所需的拓扑文件及力场参数,包括参数化配体、蛋白质、水溶液及离子等组分。
- Equilibration Simulation:通过一系列分子动力学模拟步骤使体系逐步达到热力学平衡。这些步骤包括能量最小化、分阶段升温以及恒压恒温(NPT)模拟,确保后续自由能计算的可靠性。
- Alchemy Morph:根据自由能计算路径设计配体在不同物理状态之间的变换。
- Alchemy Simulation:在一系列预定义的λ 状态下执行炼金术模拟,计算不同状态间的自由能变化。通过逐步调整分子间相互作用,平滑整个状态跃迁过程。
- Alchemy Analysis:基于炼金术模拟生成的采样数据,利用统计力学方法(MBAR、TI )计算自由能变化(∆G)。同时通过评估误差和收敛性,验证计算结果的准确性。
- 计算参考示例:
🔗GitHub:(https://github.com/freeenergylab/FEP-SPell-ABFE/tree/main/testing)
此测试对应为工作流的用法示例。本例中将会对与受体蛋白181L结合的苯和苯酚两个配体分子分别进行ABFE计算。文件夹结构如下:

用户可以将此目录复制到新的工作目录下,同时根据当前软件包设置相关环境变量以适应计算环境(参考submitBFE.sh文件中的设置用户定义变量部分),并且在config.yaml文件中修改slurm控制参数,相关内容可以进一步参考GitHub中README.md文件进行修改适配。提交作业相关命令如下:

当所有模块的任务成功结束,就会在工作目录中生成几个中间文件夹(以下划线 “_”开头)和最终结果文件dG_results.csv和dG_components_results.csv。dG_results.csv文件为预测配体的绝对结合自由能数值,dG_components_results.csv列出了每个配体分子在各个热力学变换步骤中的自由能变化值。
📊 验证结果:高精度自由能计算
FEP-SPell-ABFE 已在多个蛋白-配体体系上进行了基准测试,并与实验数据对比,表现出极高的预测准确性。
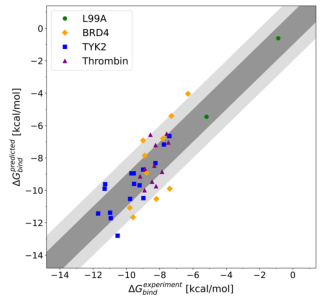
数据表明,FEP-SPell-ABFE 计算出的结合自由能与实验测量数据高度一致,具有较强的预测精度,表明其在药物筛选中的应用潜力:
- L99A(中性配体分子):RMSE 27 kcal/mol,Pearson 相关系数 1.00。
- BRD4(中性配体分子):RMSE 1.81 kcal/mol,Pearson 相关系数 0.71。
- TYK2(中性配体分子):RMSE 0.99 kcal/mol,Pearson 相关系数 0.78。
- Thrombin(+1价配体分子):RMSE 1.04 kcal/mol,Pearson 相关系数 0.53。
- 开源共享
- GitHub: https://github.com/freeenergylab/FEP-SPell-ABFE,采用 MIT 许可,免费使用
- Benchmark:https://github.com/freeenergylab/FEP-SPell-ABFE-benchmark


